
El texto detalla el desarrollo y las capacidades de AlphaFold 3, un revolucionario modelo de inteligencia artificial diseñado para predecir las estructuras de sistemas biomoleculares complejos. A diferencia de sus predecesores, esta versión utiliza una arquitectura basada en la difusión para modelar las interacciones entre proteínas, ADN, ARN, ligandos y diversas modificaciones químicas dentro de un único marco. Al sustituir los módulos antiguos por un Pairformer y un proceso de difusión generativa, el sistema alcanza una precisión sin precedentes que a menudo supera a las herramientas especializadas en el descubrimiento de fármacos y la biología molecular. El modelo gestiona eficazmente casi todos los tipos moleculares presentes en el Banco de Datos de Proteínas, proporcionando información crucial sobre las funciones celulares mediante coordenadas atómicas de alta fidelidad. A pesar de su éxito, los autores señalan limitaciones como errores estereoquímicos ocasionales y la tendencia a producir estructuras "alucinadas" en regiones desordenadas. En definitiva, la investigación demuestra que un enfoque unificado de aprendizaje profundo puede dominar la inmensa complejidad de todo el espacio biomolecular.
Enlace al artículo científico, para aquellos que quieran profundizar en el tema: "Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3", por Josh Abramson y colegas. Publicdo el 27 de Noviembre del 2024 en Nature.
El resumen, la transcripción, la traducción, y las voces fueron hechas usando herramientas de software de Inteligencia Artificial.
El resumen se presenta en la forma de un diálogo entre dos personajes sintéticos que llamaremos Alicia y Beto.
Escúchalo aquí, mientras lees la transcripción (abajo):
Transcripción
Alicia
Bienvenidos a un nuevo análisis profundo. Hoy analizamos un artículo que está causando olas verdaderamente enormes en la biología computacional.
Beto
Sí, de verdad.
Alicia
Es un lanzamiento de mayo de 2024 de Google DeepMind e Isomorphic Labs que detalla AlphaFold 3. Vamos a explorar la mecánica de esta investigación para entender por qué esta nueva arquitectura representa un atajo sin precedentes para mapear las interacciones atómicas de la vida.
Beto
Es, en efecto, un salto enorme.
Alicia
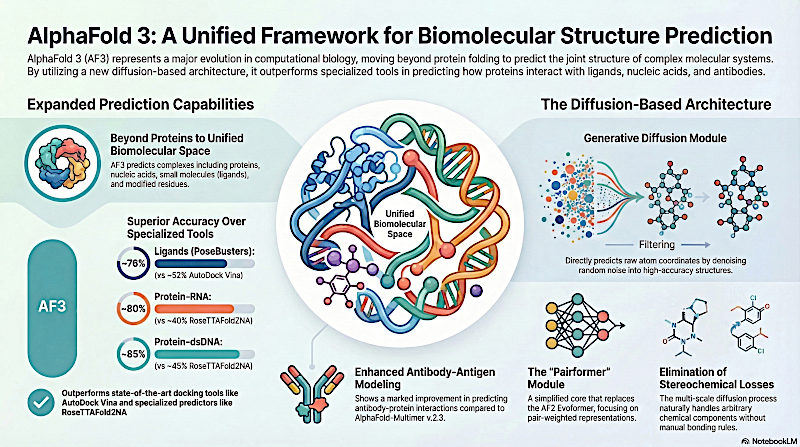
Si sigues el campo, ya conoces el legado de AlphaFold 2, que resolvió gran parte del problema del plegamiento de proteínas. Pero esta nueva iteración cambia el paradigma por completo al modelar todo el espacio biomolecular dentro de un único marco.
Bien, vamos a desglosarlo. Para comprender plenamente el salto arquitectónico, necesitamos ver lo fragmentado que estaba el panorama antes de la publicación.
Beto
El campo históricamente ha operado en estos silos altamente especializados. Tenías herramientas dedicadas para la predicción de estructuras proteicas y algoritmos completamente separados y muy afinados para, por ejemplo, el acoplamiento proteína-ligando o las interacciones con ácidos nucleicos.
Alicia
Herramientas totalmente distintas para trabajos distintos.
Beto
Exacto. Y AlphaFold 3 disuelve esas fronteras. Aporta una única arquitectura de aprendizaje profundo continua capaz de predecir la estructura conjunta de ensamblajes complejos de una sola vez: proteínas, ADN, ARN, ligandos, iones e incluso residuos modificados como la glicosilación.
Alicia
Todo manejado de forma nativa.
Beto
Todo manejado de forma nativa sin cambiar de modelo.
AlphaFold 3
Alicia
Sí. Es un enfoque unificado. Y lo que sobresale cuando se examinan los materiales suplementarios es que lograr esta universalidad requirió en realidad despojar mucha de la complejidad específica de dominio que hacía funcionar a AlphaFold 2.
Beto
Lo evolucionaron por reducción, esencialmente.
Alicia
La fuerte dependencia en los alineamientos múltiples de secuencias (MSA), que impulsaban el antiguo bloque Evoformer, se reduce de forma masiva.
Beto
El cambio del Evoformer al nuevo módulo pair‑former es una pieza de ingeniería brillante. El pair‑former sigue usando 48 bloques, pero los datos de MSA se descartan esencialmente después de la fase inicial de embedding.
Alicia
Correcto.
Beto
Esto pasa la información evolutiva condensada a una representación por pares y luego se aparta.
Alicia
Lo cual es alucinante, ¿no? Porque ¿por qué la repentina disminución de la importancia de las MSA? AlphaFold 2 prácticamente vivía y respiraba con alineamientos evolutivos profundos para inferir proximidad espacial.
Beto
Porque la generación de la estructura en el back-end del modelo es completamente diferente ahora. El modelo de difusión al final de AlphaFold 3 es increíblemente competente aprendiendo la física cruda y la estereoquímica del posicionamiento atómico directamente desde las coordenadas.
Alicia
Así que no necesita tanto el apoyo evolutivo.
Beto
Exacto. No necesita apoyarse tan fuertemente en la historia evolutiva para conjeturar la proximidad 3D de residuos, porque ya posee un entendimiento profundamente aprendido de cómo los átomos se empaquetan físicamente en el espacio.
Alicia
Y eso nos lleva al núcleo del artículo: el módulo de difusión. Los investigadores eliminaron por completo el complejo módulo de estructura de AlphaFold 2 —el que usaba atención invariante a puntos y cálculos intrincados de ángulos de torsión— y lo reemplazaron por un modelo de difusión que opera directamente sobre coordenadas cartesianas crudas.
Beto
Sí, es un cambio conceptual enorme.
Alicia
Para visualizarlo: imagina sintonizar una radio antigua.
Beto
Me gusta esa analogía.
Alicia
Empiezas con pura estática caótica y, al girar el dial, la estática se resuelve en una canción nítida. La red parte de una nube de átomos aleatorizados y les remueve el ruido, paso a paso, hasta formar un complejo biológico muy específico.
Beto
Lo fascinante es cómo ese proceso de depuración está escalonado a través de diferentes escalas. El nivel de ruido dicta las restricciones físicas que la red se ve forzada a aprender.
Alicia
¿Cómo es eso?
Beto
Pues cuando el modelo observa coordenadas muy ruidosas y fuertemente mezcladas, tiene que ignorar los detalles locales y averiguar la topología global a gran escala del complejo.
Alicia
Solo la forma aproximada.
Beto
Exacto. Y conforme el nivel de ruido baja, su foco cambia. En niveles bajos de ruido, se obsesiona con los detalles finos, perfeccionando la estereoquímica local y las geometrías de enlace.
Alicia
Y para quien nos escucha, el impacto biológico práctico de este cambio a difusión es enorme. AlphaFold 3 maneja la complejidad desordenada de compuestos químicos generales sin necesitar las penalizaciones estereoquímicas artificiales y muy afinadas que atormentaban a los modelos antiguos.
Beto
Correcto. Las penalizaciones antiguas eran muy rígidas.
Alicia
Sí, AlphaFold 2 tenía que ser explícitamente penalizado matemáticamente durante el entrenamiento si generaba un enlace que rompía las reglas de la química. AlphaFold 3 aprende desde abajo hacia arriba. Al depurar coordenadas crudas, trata un aminoácido estándar y una molécula pequeña farmacológica extraña exactamente igual: son coordenadas físicas a resolver.
Beto
Al evitar esas parametrizaciones rígidas basadas en torsiones, acomoda de manera fluida componentes químicos arbitrarios. No está atado a un diccionario predefinido de partes moleculares. Y de hecho vemos que este enfoque físico rinde dramáticamente cuando se lo compara con herramientas de docking antiguas y especializadas.
Alicia
Entremos en esos benchmarks. El artículo compara AlphaFold 3 contra el conjunto de datos Post‑Buster, enfocándose específicamente en 428 estructuras proteína‑ligando publicadas en el Protein Data Bank en 2021 o después.
Beto
Ese corte de 2021 es crítico.
Alicia
Exacto. Para prevenir filtrado de datos, asegurando que el modelo esté prediciendo estructuras novedosas en lugar de simplemente regurgitar datos de entrenamiento.
Beto
El panorama competitivo del docking ha estado dominado por herramientas clásicas como Vina o Gold. Sin embargo, esas herramientas clásicas dependen mucho de información privilegiada.
Alicia
Necesitan una ventaja inicial.
Beto
Sí. Se les alimenta con la estructura conocida y resuelta de la proteína objetivo, o al menos con las coordenadas específicas del bolsillo de unión. Su único trabajo es calcular cómo encaja el ligando en ese espacio predefinido.
Alicia
Lo cual es bastante artificial.
Beto
Demasiado. Y en un pipeline real de descubrimiento de fármacos, contar con esa estructura perfecta es un lujo que rara vez tienes.
Alicia
Pero AlphaFold 3 ejecuta un verdadero docking a ciegas. Recibe la secuencia de la proteína y la cadena SMILES del ligando, y tiene que plegar la proteína y colocar el ligando simultáneamente.
Beto
Sí.
Alicia
A pesar de la falta de entrada estructural privilegiada, AlphaFold 3 supera con creces a Vina. También aplasta a recientes herramientas de docking a ciegas basadas en aprendizaje profundo como RoseTTAFold2NA y otros.
Beto
La naturaleza generalista de la arquitectura es innegable a través de los conjuntos de datos. Al predecir interacciones proteína‑ácido nucleico, supera a RoseTTAFold2NA. Logra un rendimiento promedio mayor que las mejores presentaciones de IA en la reciente competencia CASP/CASP15 para objetivos de ARN.
Alicia
Vaya.
Beto
Y para interacciones proteína‑proteína, específicamente complejos anticuerpo‑antígeno, muestra mejoras masivas sobre AlphaFold 2.3.
Alicia
Aquí es donde se pone realmente interesante. Mirar las tasas brutas de éxito es una cosa, pero las estructuras específicas que produce este modelo destacan la escala de los cálculos. Dos ejemplos en las figuras suplementarias resaltan.
Beto
Vamos a escucharlos.
Alicia
Primero, la proteína Spike del coronavirus humano OC‑43. Hablamos de un complejo enorme de 4.665 residuos. Está fuertemente glicosilada, y el modelo la predice unida por anticuerpos neutralizantes.
Beto
Predice toda la masa del ensamblaje, incluyendo las complejas cadenas de carbohidratos y la respuesta del sistema inmune en una sola pasada.
Alicia
Simplemente increíble.
Beto
El segundo ejemplo notable es un regulador transcripcional de la familia CRP/FNR bacteriana. AlphaFold 3 predice correctamente la estructura proteica ligada a una molécula de ADN de doble cadena, mientras coloca simultáneamente un ligando cíclico GMP en su bolsillo de unión correcto.
Alicia
Ves proteína‑ADN y proteína‑ligando modeladas perfectamente al unísono. La coordinación requerida para poner todas esas posiciones atómicas bien sin choques físicos severos es un testamento al proceso de depurado multiescala.
Beto
Realmente lo es.
Alicia
Si un investigador farmacéutico puede modelar con precisión cómo una proteína objetivo interactúa con ADN, mientras visualiza simultáneamente la colocación exacta de una posible molécula terapéutica, elimina una enorme cantidad de fricción del pipeline de descubrimiento.
Beto
Lo hace. Pero apoyarse en un modelo generativo de difusión introduce nuevas clases de errores que no estaban presentes en arquitecturas previas. El más prominente es la alucinación de estructura en regiones intrínsecamente desordenadas.
Alicia
Claro. Si estás familiarizado con la IA generativa, sabes que le cuesta el concepto de la nada. En biología estructural, muchas proteínas tienen largas colas o bucles no estructurados que no adoptan una forma 3D fija a menos que interactúen con un compañero de unión.
Beto
Exacto. AlphaFold 2 era en realidad bastante bueno representando esto: generaba bucles extendidos tipo cinta indicando desorden.
Alicia
Pero la difusión funciona de manera diferente.
Beto
Correcto. El módulo de difusión crudo en AlphaFold 3 quiere naturalmente empaquetar los átomos de forma ordenada, creando estructuras densas y plausibles que son completamente ficticias.
Alicia
¿Y cómo lo arreglaron?
Beto
Para contrarrestarlo, los investigadores emplearon un método llamado cross‑distillation (destilación cruzada). Enriquecieron los datos de entrenamiento de AlphaFold 3 con predicciones estructurales generadas por la versión anterior, AlphaFold 2.3.
Alicia
El modelo antiguo.
Beto
Sí, como el modelo anterior producía de forma fiable esos bucles extendidos en regiones no estructuradas, usaron sus salidas para enseñar al nuevo modelo de difusión a imitar ese comportamiento específico, lo que redujo significativamente la tasa de alucinaciones.
Alicia
Espera: si AlphaFold 3 es fundamentalmente una arquitectura superior que entiende mejor la física del emplazamiento atómico, ¿no es contradictorio usar el modelo antiguo e inferior para entrenarlo? ¿Cómo enseña la destilación cruzada selectivamente sobre los bucles desordenados sin transferir las inexactitudes del modelo antiguo en ensamblajes complejos?
Beto
Es una gran pregunta. Se reduce a la mezcla de los datos de entrenamiento. La red se entrena con una mezcla de estructuras experimentales de alta calidad del PDB y las predicciones destiladas.
Alicia
Bien, una mezcla.
Beto
Exacto. Los datos experimentales anclan al modelo en una química estereoquímica precisa y de alta resolución para dominios plegados, mientras que los datos destilados proporcionan una señal de aprendizaje fuerte para las vastas regiones flexibles que a menudo están invisibles o faltan en los archivos experimentales del PDB. Es un parche pragmático para el sesgo generativo de la difusión.
Alicia
Lo que naturalmente conduce a la pregunta de la fiabilidad. Si el modelo está parcheando sus sesgos generativos, ¿cómo evaluamos nuestra confianza en una predicción dada?
Beto
El equipo utiliza tres métricas de confianza principales: la puntuación pLDDT predice la precisión local de átomos individuales; la PAE (Predicted Aligned Error) evalúa la confianza en las posiciones relativas de diferentes dominios; y una nueva para los complejos multi‑componente masivos que AlphaFold 3 apunta: la PDE, o matriz de error de distancia. La PDE predice específicamente el error en la distancia entre cualquier par de átomos en todo el complejo. Esto es crucial al evaluar si un ligando está realmente acoplado o simplemente flotando cerca.
Alicia
Pero entrenar esas métricas de confianza presenta un desafío matemático único en un marco de difusión. En AlphaFold 2, el modelo predecía la estructura completa de una vez, así que durante el entrenamiento podías comparar fácilmente la salida final con la verdad de referencia y entrenar la cabeza de confianza basada en ese delta. Pero el entrenamiento por difusión solo optimiza un paso aislado de la trayectoria de depuración.
Beto
Para resolver esto implementaron un procedimiento de múltiples rollouts durante el entrenamiento. No podían retropropagar a través de un rollout de la dinámica de Langevin de 1000 pasos por limitaciones de memoria. Así que ocasionalmente dejaron que el modelo ejecutara una versión rápida y de grano grueso de cuatro pasos de la generación de estructura.
Alicia
Muy ingenioso.
Beto
Sí. Toman esa estructura final predicha rápidamente y la comparan con la verdad de referencia y usan esa comparación únicamente para calibrar las cabezas pLDDT, PAE y PDE. Esto asegura que las puntuaciones de confianza reflejen la fiabilidad de la forma 3D final, no solo de un paso aislado de depuración.
Alicia
Es una solución de ingeniería muy elegante.
Pero parte de analizar críticamente este artículo requiere mirar dónde la arquitectura todavía falla. La difusión multiescala elimina en gran medida la necesidad de reglas rígidas, pero introduce peculiaridades estereoquímicas distintas. La tasa de violación de quiralidad es una enorme. El artículo señala una tasa de violación del 4,4 % en el benchmark PoseBuster.
Beto
La quiralidad es un concepto notoriamente difícil para modelos de difusión basados en coordenadas. Muchas biomoléculas existen en formas levógiras o dextrógiras; una estructura en espejo a menudo tiene exactamente las mismas distancias y ángulos locales que la correcta, convirtiéndose en un mínimo local cómodo en el paisaje energético.
Alicia
Se ve bien para el motor físico, aunque biológicamente esté invertida.
Beto
Precisamente. Así que incluso cuando AlphaFold 3 recibe explícitamente las entradas quirales correctas, el proceso espacial de depuración ocasionalmente cae en la conformación espejada.
Alicia
Cuando un centro quiral invertido puede convertir un fármaco terapéutico en una toxina, una tasa de error del 4,4 % es una realidad dura con la que los investigadores deben contar. Y los desafíos espaciales escalan con el tamaño del complejo. El artículo detalla problemas significativos con choques atómicos y estructuras masivas.
Beto
El modelo tiene problemas con solapamientos espaciales al generar ensamblajes proteína‑ácido nucleico que exceden 100 nucleótidos y 2.000 residuos. También se ve esto en casos extremos con grandes complejos homoméricos formados por múltiples cadenas proteicas idénticas, donde cadenas enteras a veces se generan ocupando exactamente el mismo espacio físico.
Alicia
Fusionadas entre sí.
Beto
Exacto. Aunque usan penalizaciones de ranking para filtrar a los peores durante la inferencia, el módulo de difusión subyacente todavía los genera.
Alicia
Eso pinta una imagen clara: cuanto mayor el sistema, mayor la probabilidad de que el proceso de difusión pliegue el espacio de coordenadas sobre sí mismo.
Pero más allá de los choques espaciales y la quiralidad, existe una limitación mucho más amplia relativa a cómo el modelo interpreta estados biológicos.
Beto
Esto plantea una pregunta importante sobre la naturaleza de los datos que consumen estos modelos. AlphaFold 3 predice una instantánea estática de alta resolución. Pero las moléculas en una célula son un conjunto dinámico. Respiran, se desplazan y alteran radicalmente su conformación dependiendo de sus socios de unión y del entorno.
Alicia
El artículo lo subraya explícitamente con una enzima llamada una ligasa E3 ubiquitina. En un entorno celular natural, esta ligasa yace en una conformación abierta cuando está vacía, el estado apo. Cuando se une a su objetivo, se cierra en una conformación ligada, el estado holo. Pero AlphaFold 3 falla completamente en capturar esa flexibilidad dinámica.
Beto
Lo hace.
Alicia
Ya sea que le pidas predecir la estructura apo vacía o la holo ligada, casi exclusivamente predice la conformación cerrada.
Beto
Es completamente ciega al estado abierto. Esto probablemente sea un artefacto de los datos de entrenamiento. El PDB está fuertemente sesgado hacia estados cristalizados ligados porque suelen ser más estables y más fáciles de capturar experimentalmente. Además, el proceso de difusión favorece naturalmente conformaciones compactas y de menor energía.
Alicia
Es un doble golpe de sesgo.
Beto
Sí. La combinación del sesgo de la base de datos y la preferencia arquitectónica significa que el modelo lucha por representar estados transitorios o abiertos. Los investigadores todavía necesitan métodos experimentales o simulaciones profundas de dinámica molecular para entender el paisaje conformacional completo de estas máquinas.
Alicia
¿Qué implica todo esto?
Si sintetizamos la arquitectura, los benchmarks y las limitaciones, AlphaFold 3 es fundamentalmente un ariete contra los silos de la biología computacional. Al abandonar algoritmos específicos de dominio en favor de un marco unificado de difusión, logra mapear la enorme diversidad de la química biológica, desde complejos extensos de ácidos nucleicos hasta el acoplamiento de pequeñas moléculas, todo en una sola pasada. Cambia la rigidez estereoquímica absoluta de los modelos antiguos por un enfoque físico y fluido del posicionamiento atómico.
Beto
La precisión que alcanza a través de un espacio químico tan vasto es algo sin precedentes para un único modelo generalista.
Conectándolo con el panorama más amplio, representa un cambio fundamental en cómo abordamos la regulación celular y el diseño terapéutico racional. Nos estamos moviendo hacia un paradigma donde modelar el entorno multicomponente completo de un objetivo farmacológico es un cálculo de base, no una búsqueda experimental de años.
Alicia
Y eso nos deja con un pensamiento final mientras digieres esta investigación. Ahora tenemos un marco unificado que puede predecir rápidamente la estructura física estática de casi cualquier combinación molecular en el cuerpo humano. Tenemos la instantánea de alta resolución de la cerradura y la llave, pero la biología es movimiento. A medida que la capacidad de cómputo escala y los modelos de difusión evolucionan, la próxima frontera no es solo generar coordenadas estáticas, sino predecir la dimensión temporal.
Imagina una arquitectura que no solo entregue un archivo PDB, sino que genere una simulación dinámica totalmente parametrizada de los engranajes moleculares girando, flexionándose e interactuando a lo largo de la vida de una célula. Cuando transicionemos de la predicción estática a la simulación molecular temporal en tiempo real, ¿cómo alterará eso nuestro enfoque fundamental para diseñar la maquinaria de la vida?
Gracias por acompañarnos en este deep dive sobre el material fuente. Sigue cuestionando, sigue explorando y sigue aprendiendo. Nos vemos la próxima vez.



{kind=link}