
Este estudio explora la integración de los Modelos de Lenguaje de Gran Tamaño (LLM) en el campo de la bioinformática, destacando su capacidad para analizar y generar datos biológicos complejos. Los investigadores clasifican estos modelos en arquitecturas de solo codificador, solo decodificador y codificador-decodificador, explicando cómo cada una se adapta a tareas específicas como la predicción de la estructura de proteínas o el análisis de secuencias genómicas. Al revisar hitos como AlphaFold y DNABERT, el texto demuestra cómo la IA está transformando nuestra comprensión del ADN, el ARN, las proteínas y la transcriptómica unicelular. A pesar de este progreso, los autores identifican obstáculos importantes, como la escasez de datos, los altos costos computacionales y la naturaleza de "caja negra" de los modelos de aprendizaje profundo. El texto concluye abogando por el aprendizaje multimodal y los modelos híbridos de IA para cerrar la brecha entre la investigación computacional y las aplicaciones clínicas. En general, el documento proporciona una hoja de ruta integral para aprovechar los modelos básicos para avanzar en la medicina de precisión y la biología de sistemas.
Enlace al artículo científico, para aquellos interesados en profundizar en el tema: "Large Language Models in Bioinformatics: A Survey", por Zhenyu Wang y colegas. Publicado el 1 de Marzo de 2026.
El resumen, la transcripción, y la traducción fueron hechas usando herramientas de software de Inteligencia Artificial.
El resumen se presenta en la forma de un diálogo entre dos personajes sintéticos que llamaremos Alicia y Beto.
Resumen
Beto
Imagina por un segundo que la inteligencia artificial no solo entendiera los idiomas que hablamos.
Alicia
Correcto.
Beto
Imagina que no solo escribiera correos por ti o tradujera francés a inglés. ¿Pero qué pasaría si en realidad pudiera leer con fluidez, traducir e incluso escribir el código fundamental de la vida misma?
Alicia
Te lo puedes imaginar.
Beto
Sí, estamos hablando del lenguaje biológico real que está operando dentro de tus células ahora mismo.
Alicia
Es un salto conceptual enorme. Estamos pasando de una era de IA que entiende texto humano a una era de IA que puede leer y reescribir activamente los planos de los organismos vivos.
Beto
Y esa es exactamente nuestra misión para hoy. Bienvenidos a un nuevo análisis profundo. Estamos explorando una encuesta exhaustiva y masiva de 2025 titulada “Large Language Models in Bioinformatics”.
Alicia
Es un paper fantástico.
Beto
De verdad que lo es. Proviene de la colaboración de mentes destacadas de la Chinese University of Hong Kong, Peking University y varias otras instituciones líderes. Nuestro objetivo hoy es darte un atajo a la vanguardia de cómo los modelos de lenguaje grande, o LLMs, están revolucionando por completo la forma en que analizamos ADN, ARN, proteínas e incluso células individuales.
Alicia
Para entender de verdad la magnitud de lo que estos investigadores han expuesto, tenemos que establecer de inmediato una diferencia crucial.
Beto
Cuéntanos.
Alicia
Los datos biológicos son muy distintos al texto que lees en Internet.
Beto
Correcto. Porque una IA entrenada en texto de Internet se apoya en estructuras de oración reconocibles.
Alicia
Exacto. Usa puntuación, gramática, sintaxis. Pero las secuencias biológicas, esas interminables cadenas de aminoácidos o nucleótidos, son altamente variables.
Beto
No tienen puntos ni comas.
Alicia
No, no las tienen. Son increíblemente específicas y están llenas de reglas tridimensionales complejas. No puedes simplemente meter eso en una IA diseñada para texto y esperar que funcione. Requiere arquitecturas de IA totalmente nuevas para descifrarlas.
Beto
Bien, desmontemos esto porque la encuesta empieza desglosando esta gramática de la vida en tres arquitecturas de IA distintas. Si los datos biológicos son tan distintos de, digamos, una página de Wikipedia, ¿cómo construyen realmente los investigadores una IA para leerlos?
Alicia
Mecanísticamente, la encuesta categoriza estos modelos de bioinformática en tres grandes paradigmas:
- modelos solo encoder (codificador),
- solo decoder (decodificador), y
- encoder-decoder (codificador-decodificador).
Beto
Vamos a verlos uno por uno.
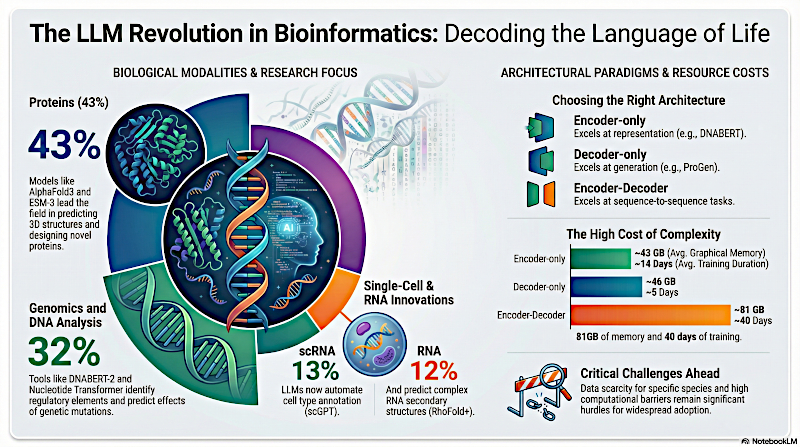
LLMs en BioInformática
Alicia
Empecemos con la arquitectura solo-encoder. Un ejemplo destacado en la encuesta es un modelo llamado ProteinBERT. La característica definitoria de un modelo solo encoder es que lee todo el contexto biológico bidireccionalmente.
Beto
Es decir, en ambas direcciones a la vez.
Alicia
Exacto. Observa la secuencia completa de izquierda a derecha y de derecha a izquierda simultáneamente.
Beto
Necesitamos una analogía: suena como hojear un párrafo entero a la vez para captar la vibra general en lugar de leer palabra por palabra.
Alicia
Es una forma perfecta de verlo, porque toma la imagen completa de una vez. Es fantástico para el aprendizaje de representaciones.
Beto
Obtiene el contexto.
Alicia
Exactamente. Captura las ricas dependencias contextuales de una secuencia de golpe. Eso lo hace brillante para tareas como la clasificación de secuencias o la identificación de un elemento regulador específico dentro de una enorme hebra de ADN.
Beto
Espera, si un modelo solo encoder ve la imagen completa de forma brillante, ¿por qué los investigadores señalan sus limitaciones en la encuesta? ¿No sería ver todo lo que hace mejor escritor si quisieras generar una nueva secuencia?
Alicia
Podrías pensarlo, pero en realidad tiene el efecto opuesto.
Beto
¿En serio?
Alicia
Sí. Porque está diseñado para procesarlo todo de un solo trago, carece del mecanismo subyacente para construir algo nuevo paso a paso. Si quieres generar una proteína completamente nueva que nunca haya existido en la naturaleza, necesitas una IA que pueda escribirla secuencialmente.
Beto
Esto nos lleva al segundo paradigma: los modelos solo-decoder.
Alicia
Precisamente.
Beto
La encuesta los etiqueta como autorregresivos. Veo que ese término se usa mucho en IA, pero ¿qué significa realmente en el contexto de la biología?
Alicia
Autorregresivo simplemente significa que el modelo genera su salida token por token, basándose completamente en la información generada previamente.
Beto
¿Como predecir la siguiente palabra?
Alicia
Sí. Piénsalo como la función de autocompletar en tu smartphone. Cuando escribes un mensaje, el teléfono adivina la palabra más probable siguiente y luego la siguiente para construir una frase.
Beto
Ah, claro.
Alicia
Un modelo solo decoder hace exactamente lo mismo, pero en lugar de palabras en inglés está prediciendo el siguiente aminoácido o el siguiente nucleótido para construir una nueva secuencia biológica.
Beto
Es literalmente un autocompletar biológico. Eso lo hace increíble para escribir textos completamente nuevos desde cero.
Alicia
Es insuperable en tareas generativas. La encuesta destaca varios modelos solo decoder, como ProGen2 y Evo, que hacen precisamente eso. Sin embargo, y aquí está la trampa: su dependencia de esta atención unidireccional paso a paso implica un enorme punto ciego.
Beto
Como solo pueden mirar hacia atrás, ...
Alicia
... les cuesta captar completamente las dependencias bidireccionales de largo alcance. En un sistema vivo, una reacción fisiológica que ocurre en un extremo de una proteína masiva puede depender mucho de una estructura situada en el otro extremo.
Beto
Un autocompletar tiene problemas para seguir la historia si el inicio del libro está 50 páginas atrás.
Alicia
Exacto. Olvida el contexto.
Beto
Así que un modelo solo-encoder lee la imagen completa de manera espléndida pero no puede escribir. Y un modelo solo-decoder es un escritor fantástico, pero le cuesta recordar el contexto a largo plazo de la historia.
Alicia
Lo entiendes muy bien.
Beto
Eso deja la tercera arquitectura que los investigadores identificaron: los modelos encoder-decoder.
Alicia
Están diseñados para tareas de secuencia a secuencia. Toman lo mejor de ambos mundos.
Beto
Entonces combinan las dos cosas.
Alicia
Correcto. Leen el contexto entero bidireccionalmente como un encoder y luego usan un decoder para traducir esa comprensión en una nueva salida. El caso de uso principal aquí es traducir una modalidad biológica a otra, como el modelo RoseTTAFold.
Beto
Mirando los datos de la encuesta, hay una gran pega en ese enfoque “lo mejor de ambos mundos” ...
Alicia
... el coste computacional.
Beto
Sí, estaba mirando las Tablas 2 y 3 del estudio. Esas tablas desglosan el coste de cómputo para estas diferentes arquitecturas, y los números son, sinceramente, difíciles de asimilar.
Alicia
Son asombrosos.
Beto
Para estos modelos encoder-decoder requieren un promedio de alrededor de 81 gigabytes de memoria gráfica por dispositivo.
Alicia
Lo cual es enorme.
Beto
Y tardan un promedio de 40 días solo para entrenar. Mi portátil se calentaría solo con abrir una docena de pestañas.
Alicia
Lo fascinante aquí es la enorme disparidad en los recursos requeridos según la arquitectura que elijas. Mencionaste el promedio de 40 días, pero los extremos son aún más salvajes.
Beto
Sí, la tabla muestra un modelo solo decoder llamado HyenaDNA que se entrenó en unos 80 minutos en una sola GPU.
Alicia
Muy manejable.
Beto
Pero luego miras un modelo encoder-decoder como xTriMOPGLM. Los investigadores anotan que tiene 100.000 millones de parámetros. Según sus datos, tardó alrededor de 150 días en entrenar, ...
Alicia
... y eso corriendo en 96 GPUs simultáneamente.
Beto
Es una cantidad de potencia de cómputo simplemente increíble.
Alicia
Lo es. Y ese coste computacional es un cuello de botella masivo para todo el campo. La complejidad computacional limita fuertemente la accesibilidad.
Beto
Claro, porque no todo el mundo tiene 96 GPUs de alta gama por ahí.
Alicia
Exacto. Si eres un grupo de investigación universitario o una startup biotecnológica pequeña con infraestructura limitada, desarrollar o incluso utilizar estos modelos encoder-decoder de última generación es increíblemente difícil.
Beto
Te deja fuera, por el precio.
Alicia
Lo hace. Obliga a la industria a buscar constantemente métodos más eficientes de compresión de modelos solo para mantener la ciencia avanzando sin necesitar una supercomputadora para cada experimento.
Beto
Es una barrera enorme, para poder entrar. Pero a pesar de ese coste, el progreso detallado en este paper es innegable.
Alicia
Absolutamente.
Beto
La encuesta incluye esta figura visual asombrosa, la figura 1. Traza todos los hitos principales de estos LLMs en los últimos años. Y mirando la gráfica circular, un contundente 43% de esos hitos de IA están enfocados puramente en proteínas.
Alicia
Casi la mitad del campo.
Beto
Sí. Ocupan la porción más grande de la tarta con diferencia.
Alicia
¿Por qué tanto del mundo de la IA está obsesionado con las proteínas?
Beto
Porque las proteínas son las máquinas físicas de la naturaleza. Hacen casi todo el trabajo pesado en tu cuerpo: digerir la comida, luchar contra virus, etc. Predecir su estructura tridimensional a partir de una secuencia plana de aminoácidos ha sido históricamente uno de los problemas más difíciles en biología. La función de una proteína está totalmente dictada por cómo se pliega en una estructura 3D.
Alicia
La forma dicta la función.
Beto
Correcto. Si no conoces la forma, no sabes qué hace.
Alicia
Y la encuesta señala modelos como AlphaFold 2 como el momento decisivo, ¿verdad? Alcanzar precisión a nivel atómico en la predicción de esas estructuras 3D.
Beto
Cambió todo.
Alicia
Pero el paper también menciona AlphaFold 3.
Beto
¿Cómo empuja eso el límite aún más?
Alicia
AlphaFold 3 va más allá de mirar una sola proteína en aislamiento. Ha avanzado hasta hacer predicciones a resolución atómica para complejos biomoleculares enteros.
Beto
¿Así que mira el sistema completo?
Alicia
Sí. Eso significa que no solo predice la forma de una máquina, sino cómo esa máquina interactúa físicamente con hebras de ADN o cómo se une con ligandos.
Beto
Para quien no recuerde, ¿qué es un ligando?
Alicia
Piensa en un ligando como un mensajero químico altamente específico. Si la proteína es una cerradura compleja, el ligando es la llave que encaja perfectamente para desencadenar una reacción biológica.
Beto
Ah, eso tiene sentido.
Alicia
Y predecir exactamente cómo un ligando encaja en una proteína es la base de la medicina moderna. Si una IA puede predecir esa interacción con precisión, puedes diseñar un fármaco en un ordenador en lugar de pasar años probando químicos al azar en un laboratorio.
Beto
Es una locura. Acabas de diseñar la llave perfecta para la cerradura.
Alicia
Exacto.
Beto
Pero predecir lo que la naturaleza ya construyó es solo el primer paso. La parte de la encuesta que realmente parece ciencia ficción es el enfoque en el diseño de novo. ¿Puedes explicar qué significa “de novo” en este contexto y qué modelos lo están haciendo?
Alicia
De novo simplemente significa desde cero. Ya no pedimos a la IA que prediga la forma de una proteína que ya existe en tu cuerpo. Le pedimos a esos modelos generativos solo decoder que inventen proteínas enteramente nuevas que la evolución nunca produjo.
Beto
Inventando biología.
Alicia
Sí. La encuesta detalla un modelo llamado ProGen2. Tiene 6.4 mil millones de parámetros y fue entrenado con más de mil millones de proteínas naturales.
Beto
Mil millones.
Alicia
Aprendiendo las reglas subyacentes de cómo encajan los aminoácidos, puede generar secuencias proteicas altamente funcionales en familias diversas sin necesidad de ajuste fino adicional.
Beto
Los investigadores también mencionan ESM-3, que adopta un enfoque multimodal, ingiriendo secuencia, estructura y datos funcionales para generar estas proteínas novedosas.
Alicia
Es una tecnología increíble.
Beto
Pero para aterrizarlo para el oyente: ¿por qué importarían las proteínas generadas desde cero en la vida real?
Alicia
Importa por las aplicaciones clínicas inmediatas. Si podemos diseñar nuevas proteínas desde cero, podemos diseñar anticuerpos personalizados con menor propensión a agregarse.
Beto
Es decir, que no se aglomeren.
Alicia
Sí. Eso los hace mucho más seguros y efectivos para los pacientes. Podemos diseñar fármacos muy específicos que se unan exactamente a proteínas malfoldadas responsables de enfermedades neurodegenerativas. Estamos pasando de observar biología a ingenierizarla para medicina de precisión.
Beto
Aquí es donde se pone realmente interesante. Si las proteínas son las máquinas 3D que hacen el trabajo en la célula, ¿de dónde obtienen sus planos?
Alicia
Del ADN.
Beto
Exacto. Y según la gráfica de la encuesta, el ADN y la genómica representan la segunda porción más grande, el 32% de los hitos encuestados.
Alicia
Entender el código genómico es la base de todo lo demás. Los modelos de IA en este espacio no solo leen una larga cadena de letras; predicen el impacto de mutaciones genéticas antes de que se manifiesten como enfermedades.
Beto
Wow.
Alicia
Identifican secuencias regulatorias ocultas que controlan cómo y cuándo se enciende o apaga un gen.
Beto
La encuesta detalla algunos pesos pesados en esta categoría. Hay una colección de modelos basados en la arquitectura BERT, como DNABERT y su sucesor DNABERT 2.
Alicia
Esos son grandes ejemplos.
Beto
Los investigadores señalan que son encoders bidireccionales pre-entrenados. Miran el contexto nucleotídico aguas arriba y aguas abajo para predecir cosas como regiones promotoras. ¿Qué es una región promotora, hablando prácticamente?
Alicia
Si un gen es un capítulo en un manual de instrucciones, una región promotora es como un enorme letrero de neón “EMPIEZA A LEER AQUÍ” colocado justo antes de que comience el capítulo.
Beto
Me gusta esa analogía.
Alicia
Le dice a la maquinaria celular exactamente dónde engancharse para empezar a copiar las instrucciones. Modelos como DinaBERT son increíblemente hábiles para encontrar estos letreros ocultos dentro de miles de millones de letras de código genético.
Beto
Eso hace que la analogía del skim-reading del encoder encaje perfectamente: está escaneando todo el genoma buscando esos letreros de neón.
Alicia
Exacto.
Beto
Pero al igual que con las proteínas, la encuesta enfatiza que el salto de entender ADN a generarlo es profundo.
Alicia
De hecho, estamos viendo modelos capaces de generar secuencias genómicas biológicamente funcionales desde cero.
Beto
Modelos como megaDNA y The Nucleotide Transformer.
Alicia
Sí, y la encuesta destaca un modelo fundacional innovador llamado Evo.
Beto
Evo.
Alicia
Lo que hace a Evo tan significativo es la escala a la que opera. Predice y genera secuencias que abarcan ADN, ARN y proteínas, yendo desde un nivel molecular pequeño hasta la escala de genoma entero.
Beto
Genera un genoma completo.
Alicia
Representa un enfoque verdaderamente holístico para el diseño biológico.
Beto
Es alucinante pensar en generar un genoma entero. Pero para obtener la imagen completa de cómo funciona una célula, tenemos que hacer zoom todavía más.
Alicia
Sí, lo tenemos que hacer.
Beto
Las porciones restantes de la gráfica son ARN con 12% y análisis de célula única con 13%. Empecemos por el ARN.
Alicia
El ARN es fascinante.
Beto
En la secundaria aprendí que el ARN era solo un mensajero pasivo: copia el plano del ADN y lo lleva a los constructores de proteínas. Pero la encuesta deja claro que el ARN es mucho más complejo que eso.
Alicia
Mucho más. Las moléculas de ARN se pliegan en estructuras secundarias y terciarias increíblemente intrincadas. Actúan como catalizadores activos. Sirven como sensores de metabolitos y regulan dinámicamente cómo se expresan los genes.
Beto
Están ocupadísimos.
Alicia
El gran reto de la IA aquí es algo que los investigadores llaman "flexibilidad conformacional".
Beto
Flexibilidad conformacional: significa que cambia físicamente su forma.
Alicia
Sí. Si el ADN es un manual de instrucciones rígido y encuadernado, el ARN es como un manual de origami que se pliega y vuelve a plegar en una herramienta funcional mientras intentas leerlo.
Beto
Eso suena imposible de leer.
Alicia
Lo es; es extremadamente difícil. Porque se está remodelando constantemente, tenemos una grave escasez de datos experimentales 3D de alta calidad para entrenar estos modelos de IA.
Beto
Así que el origami de la vida se mueve sin cesar, lo que hace la modelización tradicional una pesadilla. Pero la encuesta señala que la IA está alcanzando terreno. Modelos como RiNALMo están abordando la predicción de estructuras secundarias. Y para esas esquivas estructuras 3D, se destacan modelos como NuFold y RhoFold+ que están empezando a ofrecer predicciones de extremo a extremo directamente desde las secuencias de ARN, a pesar de la falta de datos de entrenamiento.
Alicia
Los modelos también están cartografiando cómo el ARN interactúa con otros componentes. El ARN interactúa con otras hebras de ARN, con proteínas y con pequeñas moléculas.
Beto
Y cuando esas interacciones salen mal, ...
Alicia
... a menudo llevan a enfermedades graves, incluidos varios tipos de cáncer. Por eso mapearlas con precisión con IA es una prioridad enorme para la medicina.
Beto
Una de las cosas más accesibles de toda la encuesta fue un modelo llamado RNA GPT.
Alicia
Oh, ese es brillante.
Beto
Lo describen literalmente como un modelo multimodal tipo chat para el descubrimiento de ARN. Puedes subir una secuencia de ARN y aprovecha la extensa literatura científica y modelos de lenguaje para ofrecer respuestas conversacionales concisas sobre lo que hace esa secuencia específica.
Alicia
Es esencialmente ChatGPT, pero estás conversando con una molécula en vez de con un chatbot.
Beto
Me encanta eso. Lo que nos lleva al dominio final e increíblemente complejo cubierto en la encuesta: el análisis de célula única, a menudo referido como scRNA.
Alicia
Esta es un área vital de investigación.
Beto
Creo que necesitamos otra analogía porque “análisis de célula única” suena simple, pero la encuesta lo trata como un reto computacional masivo. ¿Qué significa realmente mirar datos a nivel de célula individual?
Alicia
Piensa en la secuenciación genética tradicional en masa, como hacer un batido de frutas. Si licúas un tejido entero en un batido, puedes saborear el sabor general. Más o menos sabes qué genes están activos en ese tejido. Pero no tienes ni idea del perfil exacto de sabor de una fresa concreta o de un arándano antes de licuarlo todo.
Beto
El análisis de célula única consiste en averiguar cómo sacar esa fresa exacta y analizarla por separado.
Alicia
Exacto. Mira la expresión génica precisa de una célula específica dentro de una población enorme de millones de células.
Beto
¿Por qué es tan importante esa resolución?
Alicia
Es fundamental. Si quieres entender cómo progresa una enfermedad compleja o por qué un pequeño grupo de células cancerosas desarrolla resistencia a la quimioterapia mientras las células circundantes mueren, tienes que mirar las fresas individuales. La secuenciación en masa lo perderá por completo.
Beto
Pero el volumen de datos que eso genera debe ser astronómico. Si rastreas la expresión de miles de genes en millones de células individuales separadas, cualquier ordenador normal se colapsaría.
Alicia
El volumen de datos es asombroso, que es precisamente por qué los pipelines tradicionales ya no dan abasto. Aquí es donde los LLMs entran para asumir la carga.
Beto
Necesitan la IA solo para procesar la magnitud misma.
Alicia
Exacto. La encuesta señala modelos que operan a escala masiva solo para manejar esto. Por ejemplo, un modelo llamado scFoundation cuenta con 100 millones de parámetros y fue preentrenado con más de 50 millones de perfiles transcriptómicos humanos de célula única.
Beto
¿50 millones?
Alicia
Tiene la capacidad de manejar aproximadamente 20.000 genes simultáneamente para predecir respuestas a fármacos y anotar automáticamente diferentes tipos celulares.
Beto
El aspecto de automatización por sí solo es un cambio radical.
Alicia
Realmente lo es.
Beto
La encuesta menciona otro modelo, GPTCellType, que usa las capacidades de GPT-4 para automatizar completamente el proceso de anotación de tipos celulares, ...
Alicia
... reemplazando métodos manuales tediosos y propensos a errores que los investigadores solían tardar meses en realizar a mano.
Beto
Entonces, ¿qué significa todo esto? Hemos recorrido estas increíbles arquitecturas de IA, la enorme potencia computacional requerida y cómo estos modelos se especializan para descifrar proteínas, ADN, ARN y células individuales. Pinta un cuadro de progreso imparable.
Alicia
Lo hace, pero debemos ser realistas.
Beto
Correcto. La encuesta dedica una parte importante de su conclusión a los obstáculos que enfrentamos ahora mismo.
Alicia
Las limitaciones son muy reales y provienen principalmente de los datos mismos.
Beto
¿Cómo es eso?
Alicia
A diferencia de Internet, que proporciona un corpus abundante y prácticamente infinito de texto para entrenar LLMs tradicionales, los datos biológicos son escasos.
Beto
Y son desordenados, ¿verdad?
Alicia
Muy desordenados. A menudo son ruidosos, pueden estar incompletos y, crucialmente, los datos que sí tenemos están fuertemente sesgados.
Beto
¿Sesgados hacia qué?
Alicia
Bueno, tenemos montañas de datos sobre humanos y ratones de laboratorio comunes, pero muy poca información sobre la vasta mayoría del resto del árbol de la vida.
Beto
Ah. Entonces un modelo puede ser brillante entendiendo biología humana, pero fracasa por completo al generalizar si se le pide analizar una secuencia novedosa de una bacteria de aguas profundas o una planta rara.
Alicia
Ese es un obstáculo importante. Además, los efectos de lote y el ruido experimental de diferentes laboratorios que toman medidas distintas confunden aún más a los modelos.
Beto
Porque los datos no están estandarizados.
Alicia
Exacto. Y como comentamos con los tiempos de entrenamiento de 40 días, la barrera computacional sigue siendo inmensa. Las secuencias biológicas son increíblemente largas; procesarlas dispara los requisitos de memoria, lo que hace muy difícil aplicar arquitecturas transformer estándar a datos a escala genómica completa.
Beto
Esto plantea una pregunta importante: ¿cómo superamos estas limitaciones?
Alicia
Si lo conectamos con el panorama general, los investigadores delinean tres direcciones claras de futuro para todo el campo de la bioinformática. La primera es el empuje hacia la integración multimodal.
Beto
Porque ahora mismo, como mostró la gráfica, la mayoría de estos modelos se entrenan en silos. Solo mirando ADN o solo proteínas. Pero una célula viva no funciona aisladamente así.
Alicia
Los sistemas biológicos exhiben interacciones altamente intrincadas a través de múltiples capas moleculares simultáneamente. El futuro requiere LLMs fundacionales que puedan ingerir y procesar ADN, ARN, estructuras proteicas y epigenética todos a la vez.
Beto
Necesitamos que vean el sistema completo.
Alicia
Necesitamos arquitecturas capaces de integrar datos heterogéneos para modelar correctamente estas dependencias moleculares a varias escalas.
Beto
La segunda dirección futura que proponen los investigadores son los modelos híbridos de IA. ¿Con qué están hibridando los LMs?
Alicia
Los combinan con modelos mecanísticos, como redes neuronales de grafos (GNNs) y modelado basado en restricciones.
Beto
¿Por qué hacerlo?
Alicia
El objetivo es introducir razonamiento biológico fundamental en el proceso de aprendizaje profundo. Ahora mismo, un LLM podría generar una secuencia de proteína que parezca matemáticamente probable, pero que sea físicamente imposible de plegarse en el mundo real.
Beto
Ha alucinado una proteína que viola las leyes de la física.
Alicia
Exacto. Al hibridar la IA, los investigadores se aseguran de que las predicciones del modelo obedezcan realmente las leyes físicas de la biología y los principios evolutivos.
Beto
Básicamente le dan a la IA un control de realidad contra las leyes físicas antes de que escupa una respuesta.
Alicia
Es una gran manera de decirlo.
Beto
Y la tercera dirección que la encuesta destaca es, posiblemente, la más crucial para todos nosotros: la aplicación clínica.
Alicia
Cerrar la brecha entre una herramienta de bioinformática fascinante y una aplicación biomédica real es un reto enorme.
Beto
Una cosa es que funcione en un ordenador; otra muy distinta ponerlo en un cuerpo humano.
Alicia
Precisamente. Antes de que cualquiera de estos modelos pueda impactar de verdad la atención sanitaria cotidiana o prescribir un fármaco generado a medida, requieren evaluación clínica rigurosa y un exhaustivo benchmark experimental en laboratorios húmedos para probar su seguridad y eficacia.
Beto
Es un viaje increíble. Para sintetizar todo lo que hemos hablado hoy: somos testigos de un cambio fundamental en la capacidad humana. Pasamos de una era de simplemente leer datos biológicos con secuenciadores tradicionales a hablar activamente con ellos, usando herramientas como RNA-GPT.
Alicia
Y en última instancia a generar vida desde cero usando IA.
Beto
La encuesta de 2025 deja muy claro que la intersección entre LLMs y bioinformática está desbloqueando el código de la vida más rápido de lo que nadie predijo.
Alicia
Es un momento emocionante para estar en el campo.
Beto
Lo que nos deja con un pensamiento provocador para que lo medites hoy: hemos visto que estos modelos generativos pueden diseñar proteínas de novo y sintetizar secuencias de ADN totalmente funcionales que nunca antes existieron. Si la IA puede generar ADN y secuencias proteicas biológicamente funcionales y totalmente nuevas que la evolución natural no ideó en miles de millones de años, ¿terminará la biología convirtiéndose en otro lenguaje de programación que podamos compilar y ejecutar a demanda?
Alicia
Es una pregunta profunda, y considerando el ritmo de estos modelos, es algo con lo que la comunidad científica debatirá durante la próxima década.
Beto
Algo para que lo pienses.
Muchas gracias por acompañarnos en esta inmersión profunda en el lenguaje de la vida. Nos vemos en la próxima. Sigue explorando.



{kind=link}